Über Progeria
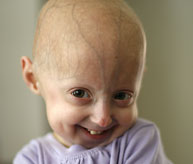 Das Hutchinson-Gilford-Progeria-Syndrom („Progeria“ oder „HGPS“) ist eine seltene, tödliche genetische Erkrankung, die durch das Auftreten eines beschleunigten Alterns bei Kindern gekennzeichnet ist. Sein Name leitet sich vom Griechischen ab und bedeutet "vorzeitig alt". Während es verschiedene Formen von Progeria * gibt, ist der klassische Typ das Hutchinson-Gilford-Progeria-Syndrom, das nach den Ärzten benannt wurde, die es erstmals in England beschrieben haben. 1886 von Dr. Jonathan Hutchinson und 1897 von Dr. Hastings Gilford.
Das Hutchinson-Gilford-Progeria-Syndrom („Progeria“ oder „HGPS“) ist eine seltene, tödliche genetische Erkrankung, die durch das Auftreten eines beschleunigten Alterns bei Kindern gekennzeichnet ist. Sein Name leitet sich vom Griechischen ab und bedeutet "vorzeitig alt". Während es verschiedene Formen von Progeria * gibt, ist der klassische Typ das Hutchinson-Gilford-Progeria-Syndrom, das nach den Ärzten benannt wurde, die es erstmals in England beschrieben haben. 1886 von Dr. Jonathan Hutchinson und 1897 von Dr. Hastings Gilford.
HGPS wird durch eine Mutation im Gen namens LMNA (ausgesprochen lamin - a) verursacht. Das LMNA-Gen produziert das Lamin A-Protein, das das Strukturgerüst darstellt, das den Zellkern zusammenhält. Forscher glauben nun, dass das defekte Lamin A-Protein den Kern instabil macht. Diese zelluläre Instabilität scheint bei Progeria zu einem vorzeitigen Alterungsprozess zu führen.
Obwohl sie gesund geboren werden, zeigen Kinder mit Progerie innerhalb der ersten zwei Lebensjahre viele Merkmale des beschleunigten Alterns. Zu den Anzeichen von Progerie gehören Wachstumsstörungen, Verlust von Körperfett und Haaren, gealtert aussehende Haut, Steifheit der Gelenke, Hüftluxation, generalisierte Atherosklerose, Herz-Kreislauf-Erkrankungen und Schlaganfall. Die Kinder sehen trotz unterschiedlicher ethnischer Herkunft bemerkenswert ähnlich aus. Ohne Behandlung sterben Kinder mit Progerie im durchschnittlichen Alter von 14.5 Jahren an Atherosklerose (Herzkrankheit).
* Andere Progeroid-Syndrome sind das Werner-Syndrom, auch als „Erwachsenen-Progerie“ bekannt, das erst in den späten Teenagerjahren mit einer Lebensspanne bis in die 40er und 50er Jahre einsetzt.
Registrieren
für unser
Newsletter!
Zusammen wir
WILL
finde das Heilmittel!
