Acerca de la progeria
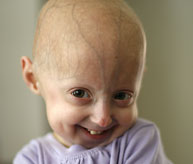 El síndrome de progeria de Hutchinson-Gilford (“progeria” o “HGPS”) es una enfermedad genética rara y mortal que se caracteriza por la aparición de un envejecimiento acelerado en los niños. Su nombre deriva del griego y significa “prematuramente viejo”. Si bien existen diferentes formas de progeria*, el tipo clásico es el síndrome de progeria de Hutchinson-Gilford, que recibió su nombre en honor a los médicos que lo describieron por primera vez en Inglaterra; en 1886 por el Dr. Jonathan Hutchinson y en 1897 por el Dr. Hastings Gilford.
El síndrome de progeria de Hutchinson-Gilford (“progeria” o “HGPS”) es una enfermedad genética rara y mortal que se caracteriza por la aparición de un envejecimiento acelerado en los niños. Su nombre deriva del griego y significa “prematuramente viejo”. Si bien existen diferentes formas de progeria*, el tipo clásico es el síndrome de progeria de Hutchinson-Gilford, que recibió su nombre en honor a los médicos que lo describieron por primera vez en Inglaterra; en 1886 por el Dr. Jonathan Hutchinson y en 1897 por el Dr. Hastings Gilford.
La HGPS es causada por una mutación en el gen llamado LMNA (pronunciado lamin – a). El gen LMNA produce la proteína Lamin A, que es el andamiaje estructural que mantiene unido el núcleo de una célula. Los investigadores ahora creen que la proteína Lamin A defectuosa hace que el núcleo sea inestable. Esa inestabilidad celular parece conducir al proceso de envejecimiento prematuro en la progeria.
Aunque nacen con un aspecto saludable, los niños con progeria comienzan a mostrar muchas características de envejecimiento acelerado durante los dos primeros años de vida. Los signos de progeria incluyen retraso del crecimiento, pérdida de grasa corporal y cabello, piel de aspecto envejecido, rigidez de las articulaciones, dislocación de cadera, aterosclerosis generalizada, enfermedad cardiovascular (cardiaca) y accidente cerebrovascular. Los niños tienen una apariencia notablemente similar, a pesar de sus diferentes orígenes étnicos. Sin tratamiento, los niños con progeria mueren de aterosclerosis (enfermedad cardíaca) a una edad promedio de 14,5 años.
* Otros síndromes progeroides incluyen el síndrome de Werner, también conocido como “progeria del adulto”, que no aparece hasta finales de la adolescencia y tiene una duración de vida hasta los 40 y 50 años.
Inscribirse
Para nuestro
¡Actualizaciones!
Juntos, nosotros
VOLUNTAD
¡Encuentra la cura!
