Vision
Our vision is a world in which every child with Progeria is cured.
Mission
To discover treatments and the cure for Progeria and its aging-related disorders, including heart disease.
Vision
Our vision is a world in which every child with Progeria is cured.
Mission
To discover treatments and the cure for Progeria and its aging-related disorders, including heart disease.
Progeria is an ultra-rare, fatal, “rapid-aging” disease that afflicts children who, without the FDA-approved treatment lonafarnib, die of heart disease at an average age of 14.5 years. PRF is the only non-profit organization solely dedicated to finding treatments and the cure for Progeria, and is making phenomenal progress toward that goal.
News

Save the Date – PRF’s 12th International Scientific Workshop
Join us at our scientific workshop taking place at the Boston Marriott Cambridge Hotel, from April 2-4, 2025, to hear about the latest breakthroughs in Progeria research.
We’re Hiring!
Join us in achieving our mission and exemplifying PRF’s core values, while making a difference in the lives of children and young adults living with Progeria around the world!

We did it – A decade of top Charity Navigator Ratings!
For the 10th straight year, PRF has earned the highest possible rating by the nation’s most trusted charity evaluator.

Global launch of PRF’s brand-new family engagement platform, Progeria Connect!
Calling all families impacted by Progeria! It’s time to get connected – to learn from each other and from PRF, share resources and experiences, and thrive as a community, no matter where in the world you live.

EXCITING NEWS – Sam Berns’ TEDx Talk Hits 100 Million Cross-Platform Views!
We are thrilled to announce that Sam Berns’ TEDx talk, ‘My Philosophy for a Happy Life,’ has now been viewed across TED and TEDx platforms more than 100 million times!

Get Involved
Our lonafarnib clinical trials enrolled 107 children from 42 different countries to test this now-FDA-approved treatment. Because of your support, these children and young adults are living longer, healthier lives.
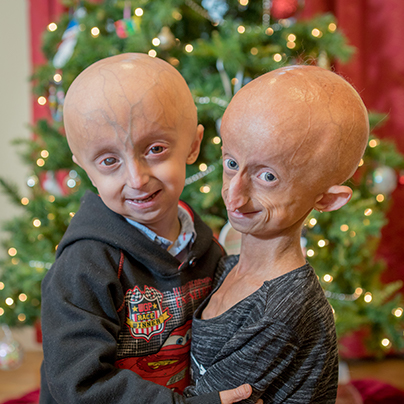
About PRF
Your donation helps The Progeria Research Foundation treat children with Progeria today, and cure them in the future.

Meet the Kids
We hope their stories inspire you to support PRF, so those dreams can come true.
Events
ONEpossible 2024
June 1- July 15
Details coming soon!
PRF’s 23rd Annual International Race for Research
September 14, 2024, Peabody, MA
Details coming soon!
ONEpossible 2024
June 1- July 15
Details coming soon!
PRF’s 23rd Annual International Race for Research
September 14, 2024, Peabody, MA
Details coming soon!
Sign Up
for Our
Newsletter!
Together, we
WILL
find the cure!
