What’s New in
Progeria Research
We’ve added this section so you can easily access information on the latest, and the most significant scientific publications on Progeria research.
In addition to the articles highlighted below, there are now hundreds of articles on Progeria and Progeria-related subjects. We suggest you search PubMed to find the specific topic(s) you are looking for.
March 2023: Exciting research milestones in treatment evaluation and life extension!
We’re excited to share two thrilling research updates with you, published online today in the world’s top cardiovascular journal, Circulation (1):
Biomarker in Progeria
A new way to measure progerin, the toxic protein that causes Progeria, has been developed by a team led by PRF co-founder and Medical Director, Dr. Leslie Gordon. With the discovery of this biomarker, which uses blood plasma to measure progerin levels, researchers can understand how treatments are affecting clinical trial participants after a shorter period of time and at multiple points along each clinical trial.
This test can optimize the clinical trial process by providing early information about the effectiveness of treatments being tested, as a lead-in to other clinical tests such as weight gain, dermatologic changes, joint contracture and function, etc., all of which require much more time to manifest. These clinical features of Progeria are important longer-term measures of treatment effects that are now complemented by the progerin levels measured earlier in therapy. We may now be able to understand treatment benefits as early as four months after starting treatment, or stop a treatment that may not benefit the trial participant, to avoid unnecessary side effects.
Even longer lives with lonafarnib
In addition to speeding up future treatment and cure discoveries, this new and innovative way to measure progerin indicates that the long-term benefit of lonafarnib for children with Progeria is greater than previously determined.
Study data indicate that lower progerin levels in the blood reflected survival benefit: the longer someone with Progeria stayed on lonafarnib, the greater the survival benefit from being on therapy. Progerin levels were decreased by about 30-60% for as long as the drug was taken, and life expectancy for patients on treatment for over 10 years was estimated to increase by almost 5 years. That’s more than a 35% increase in average lifespan, from 14.5 years to almost 20 years of age!
To learn more, see our press release here
“One of the most remarkable stories ever shared on this podcast”
– Dr. Carolyn Lam, world renowned heart specialist and host of the podcast Circulation on the Run, on the journey that led to these exciting findings. Hear the full interview about the profound impact of this study directly from Dr. Gordon. Listen here (starting at 6:41).
Hear Dr. Leslie Gordon on Circulation on the Run podcast
And in June, two editorial papers (2) and (3) were published in Circulation highlighting this biomarker’s critical importance to advancing treatments and the cure for children with Progeria and for better understanding aging.
![]()
(1) Gordon, L.B., Norris, W., Hamren, S., et al. Plasma Progerin in Patients with Hutchinson-Gilford Progeria Syndrome: Immunoassay Development and Clinical Evaluation. Circulation, 2023
(2) Progression of Cardiac Abnormalities in Hutchinson-Gilford Progeria Syndrome: A Prospective Longitudinal Study.
Olsen FJ, Gordon LB, Smoot L, Kleinman ME, Gerhard-Herman M, Hegde SM, Mukundan S, Mahoney T, Massaro J, Ha S, Prakash A. Circulation. 2023 Jun 6;147(23):1782-1784. doi: 10.1161/CIRCULATIONAHA.123.064370. Epub 2023 Jun 5.
(3) Readily Available Tools to Detect Progerin and Cardiac Disease Progression in Hutchinson-Gilford Progeria Syndrome.
Eriksson M, Haugaa K, Revêchon G. Circulation. 2023 Jun 6;147(23):1745-1747. doi: 10.1161/CIRCULATIONAHA.123.064765. Epub 2023 Jun 5.
March 2021: Exciting breakthroughs in RNA Therapeutics for Progeria!
We’re thrilled to share the results from two very exciting breakthrough studies on the use of RNA therapeutics in Progeria research. Both studies were co-funded by The Progeria Research Foundation (PRF) and co-authored by PRF’s Medical Director, Dr. Leslie Gordon.
Progerin is the disease-causing protein in Progeria. The RNA therapies interfere with the body’s ability to produce progerin, by blocking its production at the RNA level. This means that the treatment is more specific than most therapies that target progerin at the protein level.
Though each study used a different drug delivery system, both studies targeted the same basic treatment strategy, inhibiting production of RNA coding for the abnormal protein, progerin. Both were led by researchers at the National Institutes of Health (NIH), and were published today in the journal Nature Medicine.
One study, led by Francis Collins, MD, PhD, Director of the NIH, showed that treating Progeria mice with a drug named SRP2001 reduced the harmful progerin mRNA and protein expression in the aorta, the main artery in the body, as well as in other tissues. At the end of the study, the aortic wall remained stronger and the mice demonstrated an increased survival of over 60%.
“To have a targeted RNA-therapy show such significant results in an animal model gives me hope that this could lead to a major advance for the treatment of progeria,” said Collins.
The other study, led by Tom Misteli, PhD, Director of the Center for Cancer Research, National Cancer Institute, NIH, showed a 90 – 95% reduction of the toxic progerin-producing RNA in different tissues after treatment with a drug called LB143. Misteli’s lab found that progerin protein reduction was most effective in the liver, with additional improvements in the heart and aorta.
We now know there are multiple ways to decrease production of the harmful progerin protein using RNA therapeutics. Each study found different stretches of RNA in the mouse models that, when targeted, delivered an effective pathway for treatment, resulting in Progeria mice that lived much longer than those treated in previous studies with Zokinvy (lonafarnib), the only FDA approved drug for children with Progeria. Furthermore, researchers found that a combination treatment with RNA therapeutics and Zokinvy (lonafarnib) reduced progerin protein levels in liver and heart more effectively than either single treatment on its own.
“These two highly important studies demonstrate the major advancements that are now upon us in the field of targeted Progeria therapeutics,” said PRF Medical Director, Dr. Leslie Gordon. “I was thrilled to work with these brilliant research groups to advance RNA therapy for children with Progeria. Both are exciting proof-of-principle studies, and PRF is excited to forge ahead towards clinical trials that apply these treatment strategies.
—
Erdos, M.R., Cabral, W.A., Tavarez, U.L. et al. A targeted antisense therapeutic approach for Hutchinson–Gilford progeria syndrome. Nat Med (2021). https://doi.org/10.1038/s41591-021-01274-0
Puttaraju, M., Jackson, M., Klein, S. et al. Systematic screening identifies therapeutic antisense oligonucleotides for Hutchinson–Gilford progeria syndrome. Nat Med (2021). https://doi.org/10.1038/s41591-021-01262-4
January 2021: Remarkable genetic editing progress in Progeria mouse models
The science journal Nature published breakthrough results demonstrating that genetic editing in a mouse model of Progeria corrected the mutation that causes Progeria in many cells, improved several key disease symptoms and dramatically increased lifespan in the mice.
Co-funded by PRF and co-authored by PRF’s Medical Director Dr. Leslie Gordon, the study found that with a single injection of a base editor programmed to correct the disease-causing mutation, mice survived 2.5 times longer than control untreated Progeria mice, to an age corresponding with the start of old age in healthy mice. Importantly, treated mice also retained healthy vascular tissue—a significant finding, as loss of vascular integrity is a predictor of mortality in children with Progeria.
The study was co-led by world expert in genetic editing, David Liu, PhD, of the Broad Institute, MIT, Jonathan Brown, Assistant Professor of Medicine in the Division of Cardiovascular Medicine at Vanderbilt University, and Francis Collins, MD, PhD, Director of the National Institutes of Health.
“To see this dramatic response in our Progeria mouse model is one of the most exciting therapeutic developments I have been part of in 40 years as a physician-scientist,” said Dr. Collins.
“Five years ago, we were still finishing the development of the very first base editor,” said Dr. Liu. “If you had told me then that within five years, a single dose of a base editor could address Progeria in an animal at the DNA, RNA, protein, vascular pathology, and lifespan levels, I would have said ‘there’s no way.’ It’s a real testament to the dedication of the team that made this work possible.”
Additional preclinical studies are needed to investigate these results, which we hope will one day lead to a clinical trial. Read more about this exciting news in this Wall Street Journal article.
November 2020: FDA Approval for lonafarnib (Zokinvy)
On November 20, 2020, PRF achieved an important piece of our mission: lonafarnib, the first-ever treatment for Progeria, has been granted FDA approval.
Progeria now joins less than 5% of the rare diseases with an FDA-approved treatment.* Children and young adults with Progeria in the U.S. may now access lonafarnib (now called ‘Zokinvy’) by prescription, instead of through a clinical trial.
This momentous milestone has arrived thanks to 13 steadfast years of research involving four clinical trials, all co-coordinated by PRF, made possible by the courageous children and their families, and funded by you, PRF’s wonderful community of donors.
Click here for more information.
*300 rare diseases that have an FDA-approved treatment (https://www.rarediseases.info.nih.gov/diseases/FDS-orphan-drugs)/7,000 rare diseases for which the molecular basis is known (www.OMIM.org) =4.2%
April 2018: Global Study Published in JAMA Finds Treatment with Lonafarnib Extends Survival in Children with Progeria
Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome, Leslie B. Gordon, MD, PhD; Heather Shappell, PhD; Joe Massaro, PhD; Ralph B. D’Agostino Sr., PhD; Joan Brazier, MS; Susan E. Campbell, MA; Monica E. Kleinman, MD; Mark W. Kieran, MD, PhD; JAMA, April 24, 2018.
July 2016: Triple Trial Results
October 2014: PRF’s remarkable journey published in Expert Opinion
 In an article published in Expert Opinion and authored by Executive Director Audrey Gordon and Medical Director Leslie Gordon, the two PRF leaders discuss PRF’s history, goals and accomplishments, and how the PRF programs have been pivotal in the journey from obscurity to treatment.
In an article published in Expert Opinion and authored by Executive Director Audrey Gordon and Medical Director Leslie Gordon, the two PRF leaders discuss PRF’s history, goals and accomplishments, and how the PRF programs have been pivotal in the journey from obscurity to treatment.
The authors write, “It is our hope that the description of the PRF programs and services that follows, along with an account of how they are helping PRF accomplish its mission to save children with Progeria, will assist and inspire others to take similar action for the many rare disease populations that need immediate attention.”
May 2014: Study Finds Trial Medications Increase Estimated Lifespan in Children With Progeria
This study demonstrates there is evidence that a farnesyltransferase inhibitor (FTI) can extend the lives of children with Progeria by at least one-and-a-half years. The study showed an extension of mean survival of 1.6 years during the six years following initiation of treatment. Two additional drugs added later in the trials, pravastatin and zoledronate, may also contribute to this finding. This is the first evidence of treatments influencing survival for this fatal disease.
Click here for more details.
Impact of Farnesylation Inhibitors on Survival in Hutchinson-Gilford Progeria Syndrome, Leslie B. Gordon, MD, PhD, Joe Massaro, PhD, Ralph B. D’Agostino Sr., PhD, Susan E. Campbell, MA, Joan Brazier, MS, W. Ted Brown, MD, PhD, Monica E Kleinman, MD, Mark W. Kieran MD, PhD and the Progeria Clinical Trials Collaborative; Circulation, May 2, 2014 (on-line).
September 2012: First-ever Progeria Treatment for Progeria Discovered
The results of the first-ever clinical drug trial for children with Progeria reveal that Lonafarnib, a type of farnesyltransferase inhibitor (FTI) originally developed to treat cancer, has proven effective for Progeria. Every child showing improvement in one or more of four ways: gaining additional weight, better hearing, improved bone structure and/or, most importantly, increased flexibility of blood vessels. The study* was funded and coordinated by The Progeria Research Foundation.
Click here for more details.
*Gordon LB, Kleinman ME, Miller DT, Neuberg D, Giobbie-Hurder A, Gerhard-Herman M, Smoot L, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW, Clinical Trial of a Farnesyltransferase Inhibitor in Children with Hutchinson-Gilford Progeria Syndrome, Proceedings of the National Academy of Sciences, October 9, 2012 vol. 109 no. 41 16666-16671
October 2011: A Novel Approach to Progeria Therapy
That aberrant splicing in cultured skin cells of Progeria can be prevented in this manner was shown in 2005 (2). However, for treatment of patients the inhibiting reagent must be delivered intact to all tissues of the patient. it took another six years, and work in several laboratories, to develop these “delivery” methods.
In the new research (1), blocking the aberrant splicing in the model mouse resulted in impressive results. There were clear reductions in progerin concentrations in all tissues analyzed except skeletal muscle, which may have a reduced uptake of the blocking agent. The model mice recapitulated many of the phenotypes of Progeria patients, including
- Severely shortened life span (103 days compared to 2 years for wild-type mice.)
- Reduction of growth rate.
- Abnormal posture with curvature of the spine.
- Profound nuclear aberrations as a result of progerin accumulation.
- General loss of the fat layer under the skin.
- Profound bone alterations.
- Cardiovascular alterations, including significant loss of vascular smooth muscle cells.
- Alterations in the concentrations of various hormones in circulating blood plasma, including insulin and growth hormone.
The in vivo demonstration of the efficacy of reducing progerin production by blocking the aberrant splicing is a strong candidate for a valuable new approach to Progeria therapy.
(1) Osorio FG, Navarro CL, Cadiñanos J, López-Mejia IC, Quirós PM, et al, Science Translational Medicine, 3: Issue 106, advance on-line publication, October 26 (2011).
(2) Scaffidi, P. and Misteli, T. Reversal of the , cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome, Nature Medicine 11 (4): 440-445 (2005).
June 2011: PRF-funded study Identifies Rapamycin as Possible Treatment for Progeria
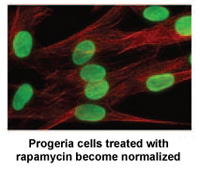
Researchers at the National Institutes of Health and Massachusetts General Hospital in Boston, MA published a new study today in Science, Translational Medicine that may lead to a new drug treatment for children with Progeria.*
Rapamycin is an FDA approved drug that has previously been shown to extend the lives of non-progeria mouse models. This new study demonstrates that rapamycin decreases the amount of the disease-causing protein progerin by 50%, improves the abnormal nuclear shape, and extends the lifespan of progeria cells. This study provides the first evidence that rapamycin may be able to decrease progerin’s damaging effects in children with progeria.
There is tremendous media coverage on this! Click below for links to media stories:
Wall Street Journal Health Blog
The Progeria Research Foundation was delighted to provide cells for this project from the PRF Cell & Tissue Bank, and help fund the research through our grants program.
This exciting new study demonstrates the remarkable pace of progeria research, while providing further insight into the aging process that affects us all.
*”Rapamycin Reverses Cellular Phenotypes and Enhances Mutant Protein Clearance in Hutchinson-Gilford Progeria Cells”
Kan Cao, John J. Graziotto, Cecilia D. Blair, Joseph R. Mazzulli, Michael R. Erdos, Dimitri Krainc, Francis S. Collins
Sci Transl Med. 2011 Jun 29;3(89):89ra58.
June 2011: Groundbreaking Study on Progeria-Aging Link
CBS Evening News, Wall Street Journal and Others Report on New Study
National Institutes of Health researchers have discovered a previously unknown link between Progeria and aging. The findings provide insights about the relationship between the toxic, Progeria-causing protein known as progerin and telomeres, which protect the ends of DNA within cells until they wear away over time and the cells die.
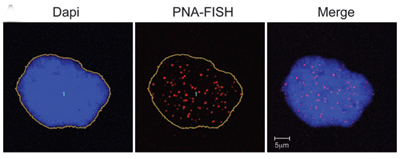
Progerin-expressing cells from normal individuals show signs of senescence.DNA in the nucleus is stained blue. Telomeres are seen as red dots.
The study* appears in the June 13, 2011 early online edition of the Journal of Clinical Investigation. It concludes that in normal aging, short or dysfunctional telomeres stimulate cells to produce progerin, which is associated with age-related cell damage.
“For the first time, we know that telomere shortening and dysfunction influences the production of progerin,” says The Progeria Research Foundation Medical Director Leslie B. Gordon, MD, PhD. “Thus these two processes, both of which influence cellular aging, are actually linked.”
Prior research has shown that progerin is not only produced in children with Progeria, but that it is produced in smaller amounts in all of us, and progerin levels increase with aging. Independently, previous research on telomere shortening and dysfunction has been associated with normal aging. Since 2003, with the discovery of the Progeria gene mutation and the progerin protein that causes the disease, one of the key areas of research has focused on understanding whether and how Progeria and aging are linked.
“Connecting this rare disease phenomenon and normal aging is bearing fruit in an important way,” said NIH Director Francis S. Collins, MD, PhD, a senior author of the paper. “This study highlights that valuable biological insights are gained by studying rare genetic disorders such as Progeria. Our sense from the start was that Progeria had a lot to teach us about the normal aging process. “
Scientists have traditionally studied telomeres and progerin separately. While there is still much to learn about whether this new connection can lead to a cure for children with Progeria or potentially be applied to extending the human lifespan, this study provides further evidence that progerin, the toxic protein discovered through finding the gene mutation in Progeria, plays a role in the normal aging process.
*Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts, Cao et al, J Clin Invest doi:10.1172/JCI43578.
Click here for the full text of the NIH press release.
May 2011: Cause of Progeroid Syndrome Discovered, Providing Further Insight into Progeria’s Link to Aging
A research team led by Progeria researcher Dr. Carlos López-Otín from the University of Oviedo in Spain encountered two families whose children have a previously unknown accelerated aging disease similar to Progeria. The children showed no defects in any genes that had previously been linked to progeroid diseases, but by studying the “coding” portions of their genomes, the team found a defect in a gene called BANF1. Family members with the progeroid disease had very low amounts of the protein made by BANF1, and, like people with Progeria, the nuclear envelopes in their cells were markedly abnormal. The abnormalities went away in cell culture experiments when the defective gene was replaced with the correct version. The findings were published in the American Journal of Human Genetics in May 2011.
BANF1 now joins the group of known genes that appear to influence some forms of premature aging—and that might affect normal aging as well.
In the past few years, scientists have been able to better understand normal aging on a molecular level thanks in part to studies of premature aging syndromes like this one as well as Progeria, which “cause the early development of characteristics normally associated with advanced age,” said López-Otín. He added that his study “underscores the importance of the nuclear lamina for human aging and demonstrates the utility of the new methods of genome sequencing to identify the genetic cause of rare and devastating diseases, which have traditionally received limited attention.”
Xose S. Puente, Victor Quesada, Fernando G. Osorio, Rubén Cabanillas, Juan Cadiñanos, Julia M. Fraile, Gonzalo R. Ordóñez, Diana A. Puente, Ana Gutiérrez-Fernández, Miriam Fanjul-Fernández et al. “Exome Sequencing and Functional Analysis Identifies BANF1 Mutation as the Cause of a Hereditary Progeroid Syndrome.” American Journal of Human Genetics, May 5, 2011 DOI: 10.1016/j.ajhg.2011.04.010
August 2010: Insulin-like Growth Factor 1 Improves Symptoms, Extends Life in a Progeroid Mouse
On August 26, 2010, Arteriosclerosis, Thrombosis, and Vascular Biology electronically published, ahead of print, the results of a study comparing Progeria and typical cardiovascular aging, entitled “Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging”. The study found that progerin, the abnormal protein that causes Progeria, is also present in the vasculature of the general population and increases with age, adding to the growing case that there are parallels between normal aging and progeria aging.
Researchers examined cardiovascular autopsies and progerin distribution in patients with Progeria along with a group without Progeria between the ages of one month and 97 years, and found that progerin in individuals without Progeria increased an average of 3.3 percent per year in the coronary arteries.
“We found similarities between many aspects of cardiovascular disease in both Progeria and the atherosclerosis that affects millions of people throughout the world” said Dr. Leslie Gordon, senior author of the study and The Progeria Research Foundation’s Medical Director. “By examining one of the rarest diseases in the world, we are gaining crucial insight into a disease that affects millions of people worldwide. Ongoing research has the potential to have a significant impact on our understanding of heart disease and aging.”
This study supports the possibility that progerin is a contributor to the risk of atherosclerosis in the general population, and merits examination as a potential new trait to help predict heart-disease risk.
Olive M, Harten I, Mitchell R, Beers J, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB.
“Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging”. Arterioscler Thromb Vasc Biol. 2010 Nov;30(11):2301-9; Epub 2010 Aug 26.
May 2010: Oxford studies show how Progeria research can further our understanding of normal aging
This situation is very similar to that in Progeria. There, prelamin A (called progerin) retains the farnesyl group. Indeed, the initial step in causing the disease is the failure to remove the farnesyl group. This failure happens because the Progeria mutation results in deletion of the part of prelamin A needed for FACE 1 to bind and remove the farnesyl group. Thus, the cause of the defects in aging and Progeria are the same: FACE1 can not do its job.
It has been known for some years that farnesyl transferase inhibitors (FTIs) inhibit (and can reverse) the presence of nuclear markers of disease in Progeria cells. Now, Shanahan et al have found that FTIs inhibit the appearance of similar nuclear markers in cells from aged normal individuals. FTIs are currently in use in Progeria clinical trials and Shanahan et al note that, these clinical trials “will shed further light on the therapeutic potential of these drugs in the treatment of aging.”
The studies described in this article are the best example to date of how studies of Progeria are furthering our understanding of normal aging.
Ragnauth CD, Warren DT, Liu Y, Shanahan CM et al, “Prelamin A Acts to Accelerate Smooth Muscle Cell Senescence and is a Novel biomarker of Human Vascular Aging.” Circulation: May 25, 2010, pp. 2200-2210.
April 2010: Further evidence that in Progeria, the presence of a farnesyl group in the progerin molecule is responsible for the disease symptoms.
Davies and coworkers prepared a new model mouse whose prelamin A, unlike RD prelamin A, is not farnesylated, but does retain the 15 amino acid sequence that is normally cleaved in the path to synthesize lamin A. This mouse does not have progeroid symptoms, indicating that in RD, as well as in Progeria, the presence of the farnesyl group, and not a change in amino acid sequence, is responsible for the disease symptoms.
DaviesBS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG,
“An accumulation of nonfarnesylated prelamin A causes cardiomyopathy but not progeria”, Hum Mol Genet. 2010 Apr 26. [Epub ahead of print]
February 2010: More Evidence FTIs provide beneficial effects through Farnesylation of Progerin
The authors evaluated the possibility that the ameliation of progeroid disease by a farnesyltransferase inhibitor (FTI) in a mouse model of Progeria is due to the effect of the drug on farnesylation of protein(s) other than progerin. They constructed a mouse that made unfarnesylated progerin, but not farnesylated progerin. This mouse also developed progeria-like disease phenotypes, but FTI did not ameliorate them. This result indicates that the drug does not act by inhibiting proteins other than progerin; it must be acting on the farnesylation of progerin, the biochemical step that is not present in the tested model.
Yang SH, Chang SY, Andres DA, Spielmann HP, Young SG, Fong LG. “Assessing the efficacy of protein farnesyltransferase inhibitors in mouse models of progeria.”
J Lipid Res. 2010 Feb;51(2):400-5. Epub 2009 Oct 26.
October 2009: The Arts Meet the Sciences in Benjamin Button Story
Maloney WJ, “Hutchinson-Gilford Progeria syndrome: its presentation in F. Scott Fitzgerald’s short story ‘the curious case of Benjamin Button’ and its oral manifestations.”
J. Dent. Res 2009 Oct 88 (10): 873-6
May 2009: Article breaks new ground on HGPS effect on cellular functions.
HGPS has previously been shown to affect many fundamental cellular functions including replication, gene expression, and DNA repair. Busch and coworkers have added the transport of proteins from the cytoplasm into the nucleus to this list. All proteins are synthesized in the cytoplasm, and those that end up being in the nucleus have to get across the nuclear membrane. The transport is accomplished through channels in the nuclear membrane called “nuclear pores”. Many proteins are too large to simply diffuse through the nuclear pores, but are “ushered” through them by special proteins that have evolved for this purpose. In this article, cells that express the mutant gene responsible for HGPS were found to have reduced transport of proteins into nuclei by direct measurement.
Busch A, Kiel T, Heupel WM, Wehnert M, Huebner S., “Nuclear protein import is reduced in cells expressing nuclear envelopathy-causing lamin A mutants.” Exp Cell Res. 2009 May 11.
April 2009: Linking Progeria and Normal Aging: Novel Insights
→ Providing structure and organization: nuclear architecture and genome integrity
→ DNA damage and repair gone awry
→ Old and beyond repair tumor suppressors and cellular senescence, and
→ Regeneration and renewal: stem-cell biology. Regeneration and renewal: stem-cell biology.
The article highlights the ways in which recent advances in the study of progeroid diseases is giving insight into basic cellular functions as well as aging.
Capell BS, Tlougan BE, Orlow SJ, “From the Rarest to the Most Common: Insights from Progeroid Syndromes into Skin Cancer and Aging.” Journal of Investigative Dermatology (2009 Apr 23), 1-11
April 2009: Past PRF Research Grantees Devise new Method to Study Progerin in Cells
Previous experiments with Fibroblast cells from Progeria patients have shown that the damage caused by the mutation is initially the result of action by the altered form of Lamin A, called Progerin. But the interpretation of these experiments can be difficult in culture for varying numbers of generations. Fong et. al. have set up an experimental system in which the amount of Progerin in Wild-type cells can be increased or decreased. This method will allow investigators to sort out the direct effects of Progerin from secondary ones, thereby advancing the study of cellular mechanisms that lead to the pathophysiology of Progeria cells.
Activating the synthesis of progerin, the mutant prelamin A in Hutchinson-Gilford progeria syndrome, with antisense oligonucleotides. (PubMed Article) Fong LG, Vickers TA, Farber EA, Choi C, Yun UJ, Hu Y, Yang SH, Coffinier C, Lee R, Yin L, Davies BS, Andres DA, Spielmann HP, Bennett CF, Young SG , “Activating the synthesis of progerin, the mutant prelamin A in Hutchinson-Gilford progeria syndrome, with antisense oligonucleotides.” Hum Mol Genet. 2009 Apr 17.
Drs. Fong and Young have previously been funded with grants from The Progeria Research Foundation.
January 2009: Quantification of Progeria Gene Expression in Normal and Progeria Cells By a New, Powerful Technique.
Swedish Team Finds a Build-up of Progerin RNA in Normal Cells as They Age
Progerin is the abnormal protein causing Progeria. In recent years, several research groups have found that normal cells also produce progerin, but much less than the cells of a child with Progeria. Moreover, the amount of progerin protein in normal cells increases as they age in the laboratory. These results established a direct link at the cellular level between Progeria and normal aging.
Dr. Maria Eriksson, author of the gene finding for Progeria in 2003, has now invented a new, powerful technique to quantitatively measure the expression of the Progeria gene. Dr. Eriksson’s laboratory at the Karolinska Institute in Sweden used the technique to measure the amount of progerin RNA in both normal and Progeria cells. RNA is the blueprint molecule in our cells for making protein. The Swedish group found that both normal and Progeria cells make larger and larger amounts of progerin RNA as they age. Eriksson’s result shows that the RNA signal for making progerin quickly builds in the cells of children with Progeria, and builds slowly over a lifetime in us all.
These new findings strengthen our understanding of the connection between normal aging and Progeria. In addition, the new technique is expected to be widely used in experiments that address the mechanism of progerin action.
Rodriguez S, Coppedè F, Sagelius H and Erikson M. “Increased expression of the Hutchinson-Gilford progeria syndrome truncated lamin A transcript during cell aging”. European Journal of Human Genetics (2009), 1-10.
August and October 2008: Is Progeria Reversible? Two recent publications show that FTIs and gene therapy may do just that!
Two separate studies show that Progeria is reversible in the cardiovascular system and the skin of mouse models. The experiments were significant in not treating the mice until they expressed Progeria symptoms, whereas most previous studies began treatment before Progeria was apparent. Production of progerin (the damaging protein made from the Progeria gene) was inhibited either by treatment with a farnesyl transferase inhibitor (FTI) or by turning off the gene. In both cases the mice reverted to normal or almost normal conditions. These observations provide encouraging evidence for the current clinical trial of FTIs for Progeria.
In a stunning display of progress with the FTI drug – now being used in the First-ever Progeria Clinical Drug Trial – Dr. Francis Collins’ research team at the National Institutes of Health * found that FTI’s prevented and even reversed the most devastating effect of Progeria in mice: cardiovascular disease.* “We were amazed that [the drug] worked so well,” says Francis Collins, a geneticist and former director of the National Human Genome Research Institute, who was senior author for the research team that identified the Progeria gene mutation in 2003. “Not only did this drug prevent these mice from developing cardiovascular disease, it reversed damage in mice that already had disease.”
The Progeria mice develop heart disease that mirrors that of children with Progeria. The authors found that the FTI was both able to prevent the development of heart disease to some degree when mice were treating from the time they were weaned, and partially reverse established disease when mice were treated beginning at age 9 months. “One of the striking things from my perspective was the ability to reverse disease, ” Collins said, which is critical given that Progeria is generally not diagnosed at birth, but only when children begin to show symptoms, when part of the damage already has been done.
“If these drugs are found to have similar effects in children, this could mark a major breakthrough for treating this devastating disease,” said NHLBI’s Dr. Nabel, who was a co-author of the study. “In addition, these findings shed light on the potential role of FTI drugs to treat other forms of coronary artery disease.”
View the article in Scientific American, “New Hope for Progeria: Drug for Rare Aging Disease”, at https://www.sciam.com/article.cfm?id=new-hope-for-progeria-drug-for-rare-aging-disease and the NIH press release at https://www.nih.gov/news/health/oct2008/nhgri-06.htm
* Capell, et. al, “A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a Progeria mouse model.” Proceedings of the National Academy of Sciences, Vol. 105, no. 41, 15902-15907 (Oct. 14, 2008)
In a second study that was published online in the Journal of Medical Genetics**, Dr. Maria Eriksson’s research team at the Karolinska Institutet in Sweden created another mouse model of Progeria with abnormalities of the skin and teeth. The mice are genetically engineered so that the Progeria mutation can be shut off at any time. Once disease was apparent, the gene for Progeria was turned off. After 13 weeks the skin was almost indistinguishable from normal skin. This study shows that in these tissues the expression of the Progeria mutation does not cause irreversible damage and that the reversal of disease is possible, which gives promise for treatment for Progeria.
**Eriksson, et. al., “Reversible phenotype in a mouse model of Hutchinson-Gilford Progeria syndrome.” J. Med. Genet. published online 15 Aug 2008; doi:10.1136/jmg.2008.060772
To purchase this article, go to: https://jmg.bmj.com/cgi/rapidpdf/jmg.2008.060772v1
More Evidence of the Link between Progeria and Normal Aging and Heart Disease
These exciting Capell and Eriksson studies show that beyond Progeria, these results have the potential to benefit all patients with cardiovascular disease. Researchers have discovered that the toxic protein responsible for Progeria is actually produced at low levels in all humans, possibly accumulating as we age. Thus, by studying these rare children, we can further our understanding of a major mechanism of human aging—and perhaps, find new ways to slow the process.
Spring 2007: Highlights of the 2007 Progeria Research Foundation Scientific Workshop: Progress in Translational Science
2006: Progeria 101/FAQ's
2004: Gene Mutation Causes Progressive Changes to Cell Structure in Children with Progeria Gene Mutation Causes Progressive Changes to Cell Structure in Children with Progeria
2003: Identification of Gene Gives Hope to Children with Progeria
Sign Up
for Our
Updates!
Together, we
WILL
find the cure!
