Was ist neu in
Progerie-Forschung
Wir haben diesen Abschnitt hinzugefügt, damit Sie einfach auf Informationen zu den neuesten und wichtigsten wissenschaftlichen Veröffentlichungen zur Progerieforschung zugreifen können.
Zusätzlich zu den unten hervorgehobenen Artikeln gibt es mittlerweile Hunderte von Artikeln zu Progerie und Progerie-bezogenen Themen. Wir empfehlen Ihnen, PubMed zu durchsuchen, um die spezifischen Themen zu finden, nach denen Sie suchen.
März 2023: Spannende Forschungsmeilensteine in der Behandlungsbewertung und Lebensverlängerung!
Wir freuen uns, Ihnen zwei spannende Forschungsupdates präsentieren zu können, die heute online im weltweit wichtigsten Fachjournal für Herz-Kreislauf-Erkrankungen veröffentlicht wurden. Auflage (1):
Biomarker bei Progerie
Ein Team unter der Leitung von PRF-Mitbegründer und medizinischer Direktor Dr. Leslie Gordon hat eine neue Methode zur Messung von Progerin entwickelt, dem toxischen Protein, das Progerie verursacht. Mit der Entdeckung dieses Biomarkers, der Blutplasma zur Messung des Progerinspiegels verwendet, Forscher können nach einer kürzeren Zeitspanne verstehen, wie sich Behandlungen auf Teilnehmer klinischer Studien auswirken und an mehreren Punkten im Verlauf jeder klinischen Studie.
Dieser Test kann den Prozess klinischer Studien optimieren, indem er Bereitstellung frühzeitiger Informationen über die Wirksamkeit getesteter Behandlungen, als Auftakt zu anderen klinischen Tests wie Gewichtszunahme, dermatologischen Veränderungen, Gelenkkontraktur und -funktion usw., die alle viel mehr Zeit benötigen, um sich zu manifestieren. Diese klinischen Merkmale der Progerie sind wichtige längerfristige Indikatoren für Behandlungseffekte, die jetzt durch die früher in der Therapie gemessenen Progerinwerte ergänzt werden. Wir können jetzt möglicherweise bereits vier Monate nach Beginn der Behandlung den Behandlungsnutzen erkennen oder eine Behandlung, die dem Studienteilnehmer möglicherweise nicht nützt, abbrechen, um unnötige Nebenwirkungen zu vermeiden.
Noch längeres Leben mit Lonafarnib
Diese neue und innovative Methode zur Messung von Progerin beschleunigt nicht nur die Entdeckung zukünftiger Behandlungsmethoden und Heilmittel, sondern weist auch darauf hin, dass die Der langfristige Nutzen von Lonafarnib für Kinder mit Progerie ist größer als bisher angenommen.
Studiendaten zeigen, dass niedrigere Progerinwerte im Blut einen Überlebensvorteil bedeuten: Je länger jemand mit Progerie Lonafarnib einnahm, desto größer war der Überlebensvorteil durch die Therapie. Die Progerinwerte sanken während der gesamten Einnahmedauer des Medikaments um etwa 30-60%, und die Lebenserwartung von Patienten, die über 10 Jahre lang behandelt wurden, erhöhte sich schätzungsweise um fast 5 Jahre. Das ist mehr als 35% Anstieg der durchschnittlichen Lebenserwartung, von 14,5 Jahren auf fast 20 Jahre!
Weitere Informationen finden Sie in unserer Pressemitteilung hier
„Eine der bemerkenswertesten Geschichten, die je in diesem Podcast erzählt wurden“
– Dr. Carolyn Lam, weltbekannte Herzspezialistin und Moderatorin des Podcasts Zirkulation auf der Flucht, über die Reise, die zu diesen spannenden Erkenntnissen führte. Hören Sie das ganze Interview über die tiefgreifende Wirkung dieser Studie direkt von Dr. Gordon. Hören Sie Hier (ab 6:41).
Hören Sie Dr. Leslie Gordon im Podcast „Circulation on the Run“
Und im Juni zwei Leitartikel (2) Und (3) erschienen in Verkehr Dies unterstreicht die entscheidende Bedeutung dieses Biomarkers für die Weiterentwicklung der Behandlung und Heilung von Kindern mit Progerie und für ein besseres Verständnis des Alterns.
![]()
(1) Gordon, LB, Norris, W., Hamren, S., et al. Plasma-Progerin bei Patienten mit Hutchinson-Gilford-Progerie-Syndrom: Entwicklung eines Immunassays und klinische Bewertung. Verkehr, 2023
(2) Fortschreiten von Herzanomalien beim Hutchinson-Gilford-Progerie-Syndrom: Eine prospektive Längsschnittstudie.
Olsen FJ, Gordon LB, Smoot L, Kleinman ME, Gerhard-Herman M, Hegde SM, Mukundan S, Mahoney T, Massaro J, Ha S, Prakash A. Verkehr. 6. Juni 2023;147(23):1782-1784. doi: 10.1161/CIRCULATIONAHA.123.064370. Epub 5. Juni 2023.
(3) Leicht verfügbare Tools zum Erkennen von Progerin und des Fortschreitens der Herzerkrankung beim Hutchinson-Gilford-Progerie-Syndrom.
Eriksson M, Haugaa K, Revêchon G. Verkehr. 6. Juni 2023;147(23):1745-1747. doi: 10.1161/CIRCULATIONAHA.123.064765. Epub 5. Juni 2023.
März 2021: Spannende Durchbrüche in der RNA-Therapeutik für Progerie!
Wir freuen uns, die Ergebnisse von zwei sehr spannende Durchbruchstudien zum Einsatz von RNA-Therapeutika in der Progerieforschung. Beide Studien wurden von der Progeria Research Foundation (PRF) mitfinanziert und von der medizinischen Direktorin der PRF, Dr. Leslie Gordon, mitverfasst.
Progerin ist das krankheitsverursachende Protein bei Progerie. Die RNA-Therapien beeinträchtigen die Fähigkeit des Körpers, Progerin zu produzieren, indem sie dessen Produktion auf RNA-Ebene blockieren. Das bedeutet, dass Die Behandlung ist spezifischer als die meisten Therapien die auf Proteinebene auf Progerin abzielen.
Obwohl in jeder Studie ein anderes Arzneimittelverabreichungssystem verwendet wurde, zielten beide Studien auf dieselbe grundlegende Behandlungsstrategie ab: die Hemmung der Produktion von RNA, die für das abnormale Protein Progerin kodiert. Beide wurden von Forschern der National Institutes of Health (NIH) geleitet und heute in der Zeitschrift veröffentlicht Naturmedizin.
Eine Studieunter der Leitung von Francis Collins, MD, PhD, Direktor des NIH, zeigte, dass die Behandlung von Progeria-Mäusen mit einem Medikament namens SRP2001 rreduzierte die schädliche Progerin-mRNA- und Proteinexpression in der Aorta, der Hauptschlagader des Körpers, sowie in anderen Geweben. Am Ende der Studie war die Aortenwand immer noch stärker und die Mäuse zeigten eine erhöhtes Überleben von über 60%.
„Dass eine gezielte RNA-Therapie in einem Tiermodell so bedeutende Ergebnisse zeigt, gibt mir Hoffnung, dass dies zu einem großen Fortschritt in der Behandlung von Progerie führen könnte“, sagte Collins.
Der andere Studieunter der Leitung von Dr. Tom Misteli, Direktor des Zentrums für Krebsforschung am National Cancer Institute der NIH, zeigte eine 90 – 95% Reduktion der toxischen Progerin-produzierenden RNA in verschiedenen Geweben nach der Behandlung mit einem Medikament namens LB143. Mistelis Labor fand heraus, dass die Reduzierung des Progerinproteins in der Leber am effektivsten war, mit zusätzlichen Verbesserungen im Herzen und in der Aorta.
Wir wissen heute, dass es mehrere Möglichkeiten gibt, die Produktion des schädlichen Progerin-Proteins mithilfe von RNA-Therapeutika zu verringern. Jede Studie fand in den Mausmodellen unterschiedliche RNA-Abschnitte, die, wenn sie gezielt angegriffen wurden, einen wirksamen Behandlungsweg lieferten, was zu Progerie-Mäuse, die viel länger lebten als die in früheren Studien mit Zokinvy (Lonafarnib) behandelten Mäuse, das einzige von der FDA zugelassene Medikament für Kinder mit Progerie. Darüber hinaus fanden Forscher heraus, dass eine Kombinationsbehandlung mit RNA-Therapeutika und Zokinvy (Lonafarnib) die Progerin-Proteinwerte in Leber und Herz wirksamer senkte als jede einzelne Behandlung für sich.
„Diese beiden äußerst wichtigen Studien zeigen die große Fortschritte, die jetzt vor uns liegen im Bereich der gezielten Progerie-Therapie“, sagte PRF Medical Director Dr. Leslie Gordon. „Ich war begeistert, mit diesen brillanten Forschungsgruppen zusammenzuarbeiten, um die RNA-Therapie für Kinder mit Progerie voranzutreiben. Beides sind spannende Proof-of-Principle-Studien, und PRF freut sich darauf, klinische Studien voranzutreiben die diese Behandlungsstrategien anwenden.
—
Erdos, MR, Cabral, WA, Tavarez, UL et al. Ein gezielter Antisense-Therapieansatz für das Hutchinson-Gilford-Progerie-Syndrom. Nat Med (2021). https://doi.org/10.1038/s41591-021-01274-0
Puttaraju, M., Jackson, M., Klein, S. et al. Durch systematisches Screening werden therapeutische Antisense-Oligonukleotide für das Hutchinson-Gilford-Progerie-Syndrom identifiziert. Nat Med (2021). https://doi.org/10.1038/s41591-021-01262-4
Januar 2021: Bemerkenswerte Fortschritte bei der genetischen Bearbeitung in Progeria-Mausmodellen
Das Wissenschaftsjournal Natur veröffentlichte bahnbrechende Ergebnisse Der Nachweis wurde erbracht, dass durch genetische Bearbeitung in einem Progerie-Mausmodell die Mutation, die in vielen Zellen Progerie verursacht, korrigiert wurde, mehrere wichtige Krankheitssymptome gelindert und die Lebensdauer der Mäuse dramatisch erhöht wurde.
Die von PRF mitfinanzierte und von PRFs medizinischem Direktor Dr. Leslie Gordon mitverfasste Studie ergab, dass Mäuse mit einer einzigen Injektion eines Baseneditors, der so programmiert war, dass er die krankheitsverursachende Mutation korrigierte, 2,5-mal länger überlebten als unbehandelte Progeria-Kontrollmäuse und ein Alter erreichten, das dem Beginn des hohen Alters bei gesunden Mäusen entspricht. Wichtig ist, dass behandelte Mäuse auch gesundes Gefäßgewebe behielten – eine bedeutende Entdeckung, da der Verlust der Gefäßintegrität ein Indikator für die Sterblichkeit bei Kindern mit Progeria ist.
Die Studie wurde gemeinsam vom weltweit führenden Experten für genetische Bearbeitung, David Liu, PhD, vom Broad Institute, MIT, Jonathan Brown, Assistenzprofessor für Medizin in der Abteilung für Herz-Kreislaufmedizin an der Vanderbilt University, und Francis Collins, MD, PhD, Direktor der National Institutes of Health, geleitet.
„Diese dramatische Reaktion in unserem Progeria-Mausmodell zu sehen, ist eine der aufregendsten therapeutischen Entwicklungen, an denen ich in meinen 40 Jahren als Arzt und Wissenschaftler beteiligt war“, sagte Dr. Collins.
„Vor fünf Jahren waren wir noch dabei, die Entwicklung des allerersten Baseneditors abzuschließen“, sagte Dr. Liu. „Hätten Sie mir damals gesagt, dass man innerhalb von fünf Jahren mit einer einzigen Dosis eines Baseneditors Progerie bei einem Tier auf DNA-, RNA-, Protein-, Gefäßpathologie- und Lebensspannenebene bekämpfen könnte, hätte ich gesagt: ‚Das ist unmöglich.‘ Es ist ein echter Beweis für das Engagement des Teams, das diese Arbeit möglich gemacht hat.“
Um diese Ergebnisse zu untersuchen, sind weitere präklinische Studien erforderlich, die hoffentlich eines Tages zu einer klinischen Studie führen werden. Lesen Sie mehr über diese spannende Neuigkeit in diesem Wall Street Journal Artikel.
November 2020: FDA-Zulassung für Lonafarnib (Zokinvy)
Am 20. November 2020 hat PRF einen wichtigen Teil unserer Mission erreicht: Lonafarnib, die allererste Behandlung gegen Progerie, hat die FDA-Zulassung erhalten.
Progerie gehört nun zu den weniger als 51 TP3T seltenen Krankheiten, für die es eine von der FDA zugelassene Behandlung gibt.* Kinder und junge Erwachsene mit Progerie können in den USA Lonafarnib (jetzt „Zokinvy“ genannt) jetzt auf Rezept und nicht mehr über eine klinische Studie beziehen.
Dieser bedeutsame Meilenstein wurde dank 13 Jahren unermüdlicher Forschung erreicht, die vier klinische Studien umfasste, die allesamt von der PRF koordiniert, durch die mutigen Kinder und ihre Familien ermöglicht und von Ihnen, der wunderbaren Spendergemeinschaft der PRF, finanziert wurden.
klicken Sie hier für weitere Informationen.
*300 seltene Krankheiten, für die es eine von der FDA zugelassene Behandlung gibt (https://www.rarediseases.info.nih.gov/diseases/FDS-orphan-drugs)/7.000 seltene Krankheiten, deren molekulare Grundlage bekannt ist (www.OMIM.org) =4,2%
April 2018: In JAMA veröffentlichte globale Studie zeigt, dass die Behandlung mit Lonafarnib das Überleben von Kindern mit Progerie verlängert
Klicken Sie hier für weitere Details.
Zusammenhang zwischen Lonafarnib-Behandlung bzw. keiner Behandlung und der Sterblichkeitsrate bei Patienten mit Hutchinson-Gilford-Progerie-Syndrom, Leslie B. Gordon, MD, PhD; Heather Shappell, PhD; Joe Massaro, PhD; Ralph B. D'Agostino Sr., PhD; Joan Brazier, MS; Susan E. Campbell, MA; Monica E. Kleinman, MD; Mark W. Kieran, MD, PhD; JAMA, 24. April 2018.
Juli 2016: Ergebnisse der Triple-Studie
Oktober 2014: PRFs bemerkenswerte Reise in Expert Opinion veröffentlicht
 In einem Artikel veröffentlicht in Expertenmeinung In dem von der Geschäftsführerin Audrey Gordon und der ärztlichen Direktorin Leslie Gordon verfassten Buch diskutieren die beiden PRF-Leiter die Geschichte, Ziele und Erfolge der PRF und wie die PRF-Programme eine entscheidende Rolle auf dem Weg aus der Bedeutungslosigkeit zur Behandlung gespielt haben.
In einem Artikel veröffentlicht in Expertenmeinung In dem von der Geschäftsführerin Audrey Gordon und der ärztlichen Direktorin Leslie Gordon verfassten Buch diskutieren die beiden PRF-Leiter die Geschichte, Ziele und Erfolge der PRF und wie die PRF-Programme eine entscheidende Rolle auf dem Weg aus der Bedeutungslosigkeit zur Behandlung gespielt haben.
Die Autoren schreiben: „Wir hoffen, dass die folgende Beschreibung der PRF-Programme und -Dienste sowie ein Bericht darüber, wie sie der PRF bei der Erfüllung ihrer Mission, an Progerie erkrankte Kinder zu retten, helfen und andere dazu inspirieren werden, ähnliche Maßnahmen für die vielen seltenen Krankheitsgruppen zu ergreifen, die sofortiger Aufmerksamkeit bedürfen.“
Mai 2014: Studie zeigt, dass Testmedikamente die geschätzte Lebenserwartung bei Kindern mit Progerie verlängern
Diese Studie zeigt, dass es Hinweise gibt, dass ein Farnesyltransferase-Hemmer (FTI) das Leben von Kindern mit Progerie um mindestens eineinhalb Jahre verlängern kann. Die Studie zeigte eine Verlängerung des mittleren Überlebens um 1,6 Jahre während der sechs Jahre nach Beginn der Behandlung. Zwei weitere Medikamente, die später in den Studien hinzugefügt wurden, Pravastatin und Zoledronat, könnten ebenfalls zu diesem Ergebnis beitragen. Dies ist der erste Beweis dafür, dass Behandlungen die Überlebenschancen bei dieser tödlichen Krankheit beeinflussen.
klicken Sie hier für weitere Details.
Auswirkungen von Farnesylierungshemmern auf das Überleben beim Hutchinson-Gilford-Progerie-Syndrom, Leslie B. Gordon, MD, PhD, Joe Massaro, PhD, Ralph B. D'Agostino Sr., PhD, Susan E. Campbell, MA, Joan Brazier, MS, W. Ted Brown, MD, PhD, Monica E Kleinman, MD, Mark W. Kieran MD, PhD und das Progeria Clinical Trials Collaborative; Verkehr, 2. Mai 2014 (online).
September 2012: Erste Behandlungsmethode für Progerie entdeckt
Die Ergebnisse von die erste klinische Arzneimittelstudie für Kinder mit Progerie zeigen, dass Lonafarnib, ein Farnesyltransferase-Hemmer (FTI), der ursprünglich zur Behandlung von Krebs entwickelt wurde, sich als wirksam gegen Progerie erwiesen hat. Jedes Kind zeigte eine Verbesserung in einer oder mehreren der folgenden vier Hinsichten: Gewichtszunahme, besseres Gehör, verbesserte Knochenstruktur und/oder, am wichtigsten, erhöhte Flexibilität der Blutgefäße. Die Studie* wurde von der Progeria Research Foundation finanziert und koordiniert.
klicken Sie hier für weitere Details.
*Gordon LB, Kleinman ME, Miller DT, Neuberg D, Giobbie-Hurder A, Gerhard-Herman M, Smoot L, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW, Klinische Studie eines Farnesyltransferase-Hemmers bei Kindern mit Hutchinson-Gilford-Progerie-Syndrom, Verfahren der Nationalen Akademie der Wissenschaften, 9. Oktober 2012, Band 109, Nr. 41, 16666-16671
Oktober 2011: Ein neuartiger Ansatz zur Progerie-Therapie
Dass auf diese Weise abnormes Spleißen in kultivierten Hautzellen von Progeria verhindert werden kann, wurde 2005 gezeigt (2). Zur Behandlung von Patienten muss das Hemmmittel jedoch intakt in alle Gewebe des Patienten transportiert werden. Es dauerte weitere sechs Jahre und die Arbeit in mehreren Labors, um diese „Transport“-Methoden zu entwickeln.
In der neuen Studie (1) führte die Blockierung des abweichenden Spleißens in der Modellmaus zu beeindruckenden Ergebnissen. Es gab deutliche Reduktionen der Progerinkonzentrationen in allen untersuchten Geweben mit Ausnahme der Skelettmuskulatur, die möglicherweise eine geringere Aufnahme des Blockierungsmittels aufweist. Die Modellmäuse rekapitulierten viele der Phänotypen von Progeriepatienten, darunter
- Stark verkürzte Lebensdauer (103 Tage im Vergleich zu 2 Jahren bei Wildtyp-Mäusen).
- Verringerung der Wachstumsrate.
- Abnorme Körperhaltung mit Verkrümmung der Wirbelsäule.
- Schwerwiegende nukleäre Aberrationen als Folge einer Progerinansammlung.
- Allgemeiner Verlust der Fettschicht unter der Haut.
- Tiefgreifende Knochenveränderungen.
- Herz-Kreislauf-Veränderungen, einschließlich eines erheblichen Verlusts an glatten Gefäßmuskelzellen.
- Veränderungen der Konzentrationen verschiedener Hormone im zirkulierenden Blutplasma, einschließlich Insulin und Wachstumshormon.
Der in vivo Der Nachweis der Wirksamkeit einer Reduzierung der Progerinproduktion durch Blockierung des abweichenden Spleißens ist ein starker Kandidat für einen wertvollen neuen Ansatz in der Progerie-Therapie.
(1) Osorio FG, Navarro CL, Cadiñanos J, López-Mejia IC, Quirós PM, et al, Science Translational Medicine, 3: Ausgabe 106, Vorabveröffentlichung im Internet, 26. Oktober (2011).
(2) Scaffidi, P. und Misteli, T. Umkehrung des zellulären Phänotyps bei der vorzeitigen Alterungskrankheit Hutchinson-Gilford-Progerie-Syndrom, Nature Medicine 11 (4): 440-445 (2005).
Juni 2011: PRF-finanzierte Studie identifiziert Rapamycin als mögliche Behandlung für Progerie
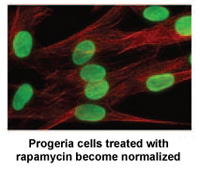
Forscher der National Institutes of Health und des Massachusetts General Hospital in Boston, MA, veröffentlichten heute eine neue Studie in Wissenschaft, Translationale Medizin das könnte zu einer neuen medikamentösen Behandlung für Kinder mit Progerie führen.*
Rapamycin ist ein von der FDA zugelassenes Medikament, das nachweislich die Lebensdauer von Mäusen ohne Progerie verlängert. Diese neue Studie zeigt, dass Rapamycin die Menge des krankheitserregenden Proteins Progerin um 50% verringert, die abnormale Kernform verbessert und die Lebensdauer von Progeriezellen verlängert. Diese Studie liefert den ersten Beweis dafür, dass Rapamycin möglicherweise die schädlichen Auswirkungen von Progerin bei Kindern mit Progerie verringern kann.
Die Medien berichten enorm darüber! Klicken Sie unten, um Links zu Medienberichten anzuzeigen:
Gesundheitsblog des Wall Street Journal
US-Nachrichten und Weltbericht
Die Progeria Research Foundation freute sich, für dieses Projekt Zellen aus der PRF Zell- und Gewebebankund helfen Sie mit, die Forschung durch unsere Stipendienprogramm.
Diese spannende neue Studie veranschaulicht das bemerkenswerte Tempo der Progerieforschung und bietet gleichzeitig weitere Einblicke in den Alterungsprozess, der uns alle betrifft.
* „Rapamycin kehrt zelluläre Phänotypen um und verbessert die Clearance mutierter Proteine in Hutchinson-Gilford-Progeriezellen“
Kan Cao, John J. Graziotto, Cecilia D. Blair, Joseph R. Mazzulli, Michael R. Erdos, Dimitri Krainc, Francis S. Collins
Sci Transl Med. 2011 Jun 29;3(89):89ra58.
Juni 2011: Bahnbrechende Studie zum Zusammenhang zwischen Progerie und Alterung
CBS Abendnachrichten, Wall Street Journal Und Sonstiges Bericht zur neuen Studie
Forscher des National Institutes of Health haben einen bisher unbekannten Zusammenhang zwischen Progerie und Alterung entdeckt. Die Ergebnisse geben Einblicke in die Beziehung zwischen dem toxischen, Progerie verursachenden Protein namens Progerin Und Telomere, die die Enden der DNA in den Zellen schützen, bis sie mit der Zeit abgenutzt werden und die Zellen absterben.
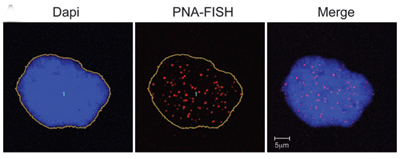
Progerin-exprimierende Zellen von normalen Individuen zeigen Anzeichen von Seneszenz. DNA im Zellkern ist blau gefärbt. Telomere sind als rote Punkte zu sehen.
Die Studie* erscheint in der Online-Frühausgabe des Journal of Clinical Investigation vom 13. Juni 2011. Sie kommt zu dem Schluss, dass bei normalem Altern kurze oder dysfunktionale Telomere die Zellen zur Produktion von Progerin anregen, das mit altersbedingten Zellschäden in Zusammenhang steht.
„„Zum ersten Mal wissen wir, dass die Verkürzung und Funktionsstörung der Telomere die Produktion von Progerin beeinflusst“, sagt Leslie B. Gordon, MD, PhD, medizinischer Direktor der Progeria Research Foundation. „Diese beiden Prozesse, die beide die Zellalterung beeinflussen, sind also tatsächlich miteinander verbunden.“
Frühere Forschungen haben gezeigt, dass Progerin nicht nur bei Kindern mit Progerie produziert wird, sondern dass es in kleineren Mengen bei uns allen vorkommt und dass der Progerinspiegel mit dem Alter steigt. Unabhängig davon wurde frühere Forschung zur Verkürzung und Funktionsstörung der Telomere mit normalem Altern in Verbindung gebracht. Seit 2003, mit der Entdeckung der Progerie-Genmutation und des Progerin-Proteins, das die Krankheit verursacht, konzentriert sich einer der wichtigsten Forschungsbereiche auf das Verständnis, ob und wie Progerie und Altern zusammenhängen.
„Die Verbindung dieses seltenen Krankheitsphänomens mit normalem Altern trägt wichtige Früchte“, sagte NIH-Direktor Francis S. Collins, MD, PhD, einer der Hauptautoren der Studie. „Diese Studie unterstreicht, dass durch die Untersuchung seltener genetischer Erkrankungen wie Progerie wertvolle biologische Erkenntnisse gewonnen werden. Wir hatten von Anfang an das Gefühl, dass Progerie uns viel über den normalen Alterungsprozess beibringen kann.“
Wissenschaftler haben Telomere und Progerin traditionell getrennt untersucht. Obwohl noch viel darüber zu lernen ist, ob diese neue Verbindung zu einer Heilung für Kinder mit Progerie führen oder möglicherweise zur Verlängerung der menschlichen Lebensspanne eingesetzt werden kann, liefert diese Studie weitere Beweise dafür, dass Progerin, das giftige Protein, das durch die Entdeckung der Genmutation bei Progerie entdeckt wurde, eine Rolle im normalen Alterungsprozess spielt.
*Progerin und Telomer-Dysfunktion wirken zusammen und lösen zelluläre Seneszenz in normalen menschlichen Fibroblasten aus, Cao et al, J Clin Invest doi:10.1172/JCI43578.
klicken Sie hier für den vollständigen Text der Pressemitteilung des NIH.
Mai 2011: Ursache des Progeroid-Syndroms entdeckt, was weitere Einblicke in den Zusammenhang zwischen Progerie und Alterung bietet
Ein Forschungsteam unter der Leitung des Progerie-Forschers Carlos López-Otín von der Universität Oviedo in Spanien trafen zwei Familien, deren Kinder an einer bisher unbekannten beschleunigten Alterungskrankheit leiden, die Progerie ähnelt. Die Kinder wiesen keine Defekte in Genen auf, die zuvor mit progeroiden Krankheiten in Verbindung gebracht worden waren, aber durch die Untersuchung der „kodierenden“ Teile ihrer Genome entdeckte das Team einen Defekt in einem Gen namens BANF1. Familienmitglieder mit der progeroiden Krankheit hatten sehr geringe Mengen des von BANF1 produzierten Proteins, und wie bei Menschen mit Progerie waren die Kernhüllen in ihren Zellen deutlich abnormal. Die Anomalien verschwanden in Zellkulturexperimenten, als das defekte Gen durch die richtige Version ersetzt wurde. Die Ergebnisse wurden in der Amerikanisches Journal der Humangenetik im Mai 2011.
BANF1 reiht sich nun in die Gruppe bekannter Gene ein, die offenbar einige Formen vorzeitiger Alterung beeinflussen – und möglicherweise auch die normale Alterung beeinflussen.
In den letzten Jahren konnten Wissenschaftler das normale Altern auf molekularer Ebene besser verstehen, unter anderem dank Studien zu vorzeitigen Alterungssyndromen wie diesem und Progerie, die „zu einer frühen Entwicklung von Merkmalen führen, die normalerweise mit fortgeschrittenem Alter in Verbindung gebracht werden“, sagte López-Otín. Er fügte hinzu, dass seine Studie „die Bedeutung der Kernlamina für das menschliche Altern unterstreicht und die Nützlichkeit der neuen Methoden der Genomsequenzierung zeigt, um die genetische Ursache seltener und verheerender Krankheiten zu identifizieren, denen traditionell nur wenig Aufmerksamkeit geschenkt wurde.“
Xose S. Puente, Victor Quesada, Fernando G. Osorio, Rubén Cabanillas, Juan Cadiñanos, Julia M. Fraile, Gonzalo R. Ordóñez, Diana A. Puente, Ana Gutiérrez-Fernández, Miriam Fanjul-Fernández et al. „Exomsequenzierung und Funktionsanalyse identifizieren BANF1-Mutation als Ursache eines hereditären Progeroid-Syndroms.“ American Journal of Human Genetics, 5. Mai 2011 DOI: 10.1016/j.ajhg.2011.04.010
August 2010: Insulinähnlicher Wachstumsfaktor 1 lindert Symptome und verlängert das Leben einer progeroiden Maus
Am 26. August 2010 Arteriosklerose, Thrombose und Gefäßbiologie Die Ergebnisse einer Studie, die Progerie und typische kardiovaskuläre Alterung vergleicht, wurden vor dem Druck elektronisch veröffentlicht. Der Titel lautete „Kardiovaskuläre Pathologie bei Hutchinson-Gilford-Progerie: Korrelation mit der vaskulären Pathologie des Alterns“. Die Studie ergab, dass Progerin, das abnormale Protein, das Progerie verursacht, auch im Gefäßsystem der Allgemeinbevölkerung vorhanden ist und mit dem Alter zunimmt, was die zunehmende Annahme untermauert, dass es Parallelen zwischen normaler Alterung und Progerie-Alterung gibt.
Forscher untersuchten Herz-Kreislauf-Autopsien und die Progerin-Verteilung bei Patienten mit Progerie zusammen mit einer Gruppe ohne Progerie im Alter zwischen einem Monat und 97 Jahren und fanden heraus, dass der Progerinspiegel in den Koronararterien bei Personen ohne Progerie jährlich um durchschnittlich 3,3 Prozent zunimmt.
„Wir haben Ähnlichkeiten zwischen vielen Aspekten der Herz-Kreislauf-Erkrankungen sowohl bei Progerie als auch bei Arteriosklerose festgestellt, von der Millionen Menschen auf der ganzen Welt betroffen sind“, sagte Dr. Leslie Gordon, Hauptautorin der Studie und medizinische Direktorin der Progeria Research Foundation. „Durch die Untersuchung einer der seltensten Krankheiten der Welt gewinnen wir entscheidende Erkenntnisse über eine Krankheit, von der Millionen Menschen weltweit betroffen sind. Laufende Forschung hat das Potenzial, unser Verständnis von Herzkrankheiten und Alterung erheblich zu beeinflussen.“
Diese Studie stützt die Annahme, dass Progerin das Arterioskleroserisiko in der Allgemeinbevölkerung erhöht, und bedarf einer näheren Untersuchung, da es als potenzielles neues Merkmal zur Vorhersage des Risikos einer Herzerkrankung herangezogen werden kann.
Olive M, Harten I, Mitchell R, Beers J, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB.
„Kardiovaskuläre Pathologie bei Hutchinson-Gilford-Progerie: Korrelation mit der vaskulären Pathologie des Alterns“. Arteriosklerose, Thrombose, Vasc. Biol. 2010 Nov;30(11):2301-9; Epub 2010, 26. August.
Mai 2010: Oxford-Studien zeigen, wie die Progerieforschung unser Verständnis des normalen Alterns erweitern kann
Diese Situation ist der bei Progerie sehr ähnlich. Dort behält Prelamin A (Progerin genannt) die Farnesylgruppe. Tatsächlich ist der erste Schritt zur Entstehung der Krankheit das Versagen bei der Entfernung der Farnesylgruppe. Dieses Versagen tritt auf, weil die Progerie-Mutation zur Deletion des Teils von Prelamin A führt, der für FACE 1 zum Binden und Entfernen der Farnesylgruppe erforderlich ist. Die Ursache der Defekte beim Altern und bei Progerie ist also dieselbe: FACE1 kann seine Aufgabe nicht erfüllen.
Es ist seit einigen Jahren bekannt, dass Farnesyltransferase-Inhibitoren (FTIs) das Auftreten von nukleären Krankheitsmarkern in Progeriezellen hemmen (und rückgängig machen können). Shanahan et al. haben nun herausgefunden, dass FTIs das Auftreten ähnlicher nukleärer Marker in Zellen von gesunden älteren Menschen hemmen. FTIs werden derzeit in klinischen Studien zu Progerie eingesetzt und Shanahan et al. weisen darauf hin, dass diese klinischen Studien „weiteres Licht auf das therapeutische Potenzial dieser Medikamente bei der Behandlung des Alterns werfen werden“.
Die in diesem Artikel beschriebenen Studien sind bislang das beste Beispiel dafür, wie Studien zur Progerie unser Verständnis des normalen Alterns erweitern.
Ragnauth CD, Warren DT, Liu Y, Shanahan CM et al., „Prelamin A beschleunigt die Alterung glatter Muskelzellen und ist ein neuer Biomarker für die Gefäßalterung beim Menschen.“ Circulation: 25. Mai 2010, S. 2200–2210.
April 2010: Weitere Beweise dafür, dass bei Progerie das Vorhandensein einer Farnesylgruppe im Progerinmolekül für die Krankheitssymptome verantwortlich ist.
Davies und seine Kollegen haben ein neues Mausmodell entwickelt, dessen Prelamin A im Gegensatz zu RD-Prelamin A nicht farnesyliert ist, das aber die 15 Aminosäuren umfassende Sequenz beibehält, die normalerweise auf dem Weg zur Synthese von Lamin A gespalten wird. Diese Maus zeigt keine progeroiden Symptome, was darauf hindeutet, dass bei RD wie auch bei Progerie die Anwesenheit der Farnesylgruppe und nicht eine Veränderung der Aminosäuresequenz für die Krankheitssymptome verantwortlich ist.
DaviesBS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG,
„Eine Ansammlung von nichtfarnesyliertem Prelamin A verursacht Kardiomyopathie, aber keine Progerie“, Hum Mol Genet. 26. April 2010. [Epub vor Drucklegung]
Februar 2010: Weitere Belege für positive Effekte von FTIs durch Farnesylierung von Progerin
Die Autoren untersuchten die Möglichkeit, dass die Linderung der Progerie-Krankheit durch einen Farnesyltransferase-Hemmer (FTI) in einem Mausmodell von Progerie auf die Wirkung des Medikaments auf die Farnesylierung anderer Proteine als Progerin zurückzuführen ist. Sie konstruierten eine Maus, die nichtfarnesyliertes Progerin, aber kein farnesyliertes Progerin produzierte. Diese Maus entwickelte ebenfalls progerieähnliche Krankheitsphänomene, die jedoch durch FTI nicht gelindert wurden. Dieses Ergebnis weist darauf hin, dass das Medikament nicht durch Hemmung anderer Proteine als Progerin wirkt; es muss auf die Farnesylierung von Progerin wirken, den biochemischen Schritt, der im getesteten Modell nicht vorhanden ist.
Yang SH, Chang SY, Andres DA, Spielmann HP, Young SG, Fong LG. „Bewertung der Wirksamkeit von Protein-Farnesyltransferase-Inhibitoren in Mausmodellen von Progerie.“
J Lipid Res. 2010 Feb;51(2):400-5. Epub 2009 Okt 26.
Oktober 2009: Die Künste treffen die Wissenschaften in Benjamin Button Story
Maloney WJ, „Hutchinson-Gilford-Progerie-Syndrom: seine Darstellung in F. Scott Fitzgeralds Kurzgeschichte ‚Der seltsame Fall des Benjamin Button‘ und seine mündlichen Manifestationen.“
J. Dent. Res 2009 Okt 88 (10): 873-6
Mai 2009: Der Artikel betritt Neuland hinsichtlich der Auswirkungen von HGPS auf Zellfunktionen.
Es wurde bereits gezeigt, dass HGPS viele grundlegende Zellfunktionen beeinflusst, darunter Replikation, Genexpression und DNA-Reparatur. Busch und seine Kollegen haben dieser Liste den Transport von Proteinen vom Zytoplasma in den Zellkern hinzugefügt. Alle Proteine werden im Zytoplasma synthetisiert, und diejenigen, die schließlich in den Zellkern gelangen, müssen die Kernmembran durchqueren. Der Transport erfolgt durch Kanäle in der Kernmembran, die als „Kernporen“ bezeichnet werden. Viele Proteine sind zu groß, um einfach durch die Kernporen zu diffundieren, sondern werden von speziellen Proteinen, die sich zu diesem Zweck entwickelt haben, durch sie „geschleust“. In diesem Artikel wurde durch direkte Messung festgestellt, dass Zellen, die das für HGPS verantwortliche mutierte Gen exprimieren, einen reduzierten Proteintransport in die Kerne aufweisen.
Busch A, Kiel T, Heupel WM, Wehnert M, Huebner S., „Der Import von Kernproteinen ist in Zellen, die Lamin-A-Mutanten exprimieren, die eine Kernenvelopathie verursachen, reduziert.“ Exp. Zellres. 11. Mai 2009.
April 2009: Zusammenhang zwischen Progerie und normalem Altern: Neue Erkenntnisse
→ Bereitstellung von Struktur und Organisation: Kernarchitektur und Genomintegrität
→ DNA-Schäden und -Reparatur schiefgelaufen
→ Alte und nicht mehr reparierbare Tumorsuppressoren und zelluläre Seneszenz und
→ Regeneration und Erneuerung: Stammzellenbiologie. Regeneration und Erneuerung: Stammzellenbiologie.
Der Artikel beleuchtet, wie die jüngsten Fortschritte in der Erforschung progeroider Erkrankungen Einblicke in grundlegende Zellfunktionen und den Alterungsprozess geben.
Capell BS, Tlougan BE, Orlow SJ, „Vom Seltensten zum Häufigsten: Erkenntnisse aus Progeroid-Syndromen zu Hautkrebs und Alterung.“ Zeitschrift für investigative Dermatologie (23. April 2009), 1-11
April 2009: Ehemalige PRF-Forschungsstipendiaten entwickeln neue Methode zur Untersuchung von Progerin in Zellen
Frühere Experimente mit Fibroblastenzellen von Progeriepatienten haben gezeigt, dass der durch die Mutation verursachte Schaden zunächst das Ergebnis der Wirkung der veränderten Form von Lamin A, genannt Progerin, ist. Die Interpretation dieser Experimente kann jedoch in Kulturen über eine unterschiedliche Anzahl von Generationen hinweg schwierig sein. Fong et al. haben ein experimentelles System entwickelt, in dem die Menge an Progerin in Wildtyp Zellen können erhöht oder verringert werden. Mit dieser Methode können Forscher die direkten Auswirkungen von Progerin von den sekundären unterscheiden und so die Erforschung der zellulären Mechanismen vorantreiben, die zur Pathophysiologie von Progeriazellen führen.
Aktivierung der Synthese von Progerin, dem mutierten Prälamin A beim Hutchinson-Gilford-Progerie-Syndrom, mit Antisense-Oligonukleotiden. (PubMed-Artikel) Fong LG, Vickers TA, Farber EA, Choi C, Yun UJ, Hu Y, Yang SH, Coffinier C, Lee R, Yin L, Davies BS, Andres DA, Spielmann HP, Bennett CF, Young SG, „Aktivierung der Synthese von Progerin, dem mutierten Prälamin A beim Hutchinson-Gilford-Progerie-Syndrom, mit Antisense-Oligonukleotiden.“ Hum Mol Genet. 17. April 2009.
Dr. Fong und Dr. Young wurden bereits zuvor mit Zuschüssen der Progeria Research Foundation gefördert.
Januar 2009: Quantifizierung der Progerie-Genexpression in normalen und Progerie-Zellen durch eine neue, leistungsfähige Technik.
Schwedisches Team entdeckt im Alter eine Ansammlung von Progerin-RNA in normalen Zellen
Progerin ist das abnormale Protein, das Progerie verursacht. In den letzten Jahren haben mehrere Forschungsgruppen herausgefunden, dass auch normale Zellen Progerin produzieren, allerdings viel weniger als die Zellen eines Kindes mit Progerie. Darüber hinaus nimmt die Menge an Progerinprotein in normalen Zellen mit zunehmendem Alter im Labor zu. Diese Ergebnisse stellten auf zellulärer Ebene eine direkte Verbindung zwischen Progerie und normalem Altern her.
Dr. Maria Eriksson, Autorin der Genentdeckung für Progerie im Jahr 2003, hat nun eine neue, leistungsstarke Technik erfunden, um die Expression des Progerie-Gens quantitativ zu messen. Dr. Erikssons Labor am Karolinska-Institut in Schweden verwendete die Technik, um die Menge an Progerin-RNA sowohl in normalen als auch in Progerie-Zellen zu messen. RNA ist das Bauplanmolekül in unseren Zellen für die Proteinherstellung. Die schwedische Gruppe fand heraus, dass sowohl normale als auch Progerie-Zellen mit zunehmendem Alter immer größere Mengen an Progerin-RNA produzieren. Erikssons Ergebnisse zeigen, dass sich das RNA-Signal zur Herstellung von Progerin in den Zellen von Kindern mit Progerie schnell aufbaut und sich bei uns allen im Laufe des Lebens langsam aufbaut.
Diese neuen Erkenntnisse stärken unser Verständnis des Zusammenhangs zwischen normalem Altern und Progerie. Darüber hinaus wird erwartet, dass die neue Technik in Experimenten, die sich mit dem Wirkmechanismus von Progerin befassen, weit verbreitet eingesetzt wird.
Rodriguez S, Coppedè F, Sagelius H und Erikson M. „Erhöhte Expression des verkürzten Lamin-A-Transkripts des Hutchinson-Gilford-Progerie-Syndroms während der Zellalterung“. Europäisches Journal der Humangenetik (2009), 1-10.
August und Oktober 2008: Ist Progerie umkehrbar? Zwei aktuelle Veröffentlichungen zeigen, dass FTIs und Gentherapie genau das können!
Zwei separate Studien zeigen, dass Progerie im Herzkreislaufsystem und in der Haut von Mäusemodellen reversibel ist. Die Experimente waren deshalb bedeutsam, weil die Mäuse erst behandelt wurden, als sie Symptome von Progerie zeigten, während die meisten früheren Studien mit der Behandlung begannen, bevor Progerie sichtbar wurde. Die Produktion von Progerin (dem schädlichen Protein, das aus dem Progerie-Gen hergestellt wird) wurde entweder durch die Behandlung mit einem Farnesyltransferase-Inhibitor (FTI) oder durch das Ausschalten des Gens gehemmt. In beiden Fällen kehrten die Mäuse in einen normalen oder fast normalen Zustand zurück. Diese Beobachtungen liefern ermutigende Beweise für die aktuelle klinische Studie von FTIs gegen Progerie.
In einem beeindruckenden Beispiel für den Fortschritt des FTI-Medikaments – das jetzt in der Erste klinische Arzneimittelstudie zu Progerie – Das Forschungsteam von Dr. Francis Collins am National Institutes of Health* stellte fest, dass FTIs die verheerendste Auswirkung von Progerie bei Mäusen verhinderten und sogar rückgängig machten: Herz-Kreislauf-Erkrankungen.* „Wir waren überrascht, dass [das Medikament] so gut wirkte“, sagt Francis Collins, ein Genetiker und ehemaliger Direktor des National Human Genome Research Institute, der leitender Autor des Forschungsteams war, das 2003 die Progerie-Genmutation entdeckte. „Dieses Medikament verhinderte nicht nur, dass diese Mäuse Herz-Kreislauf-Erkrankungen entwickelten, sondern machte auch die Schäden bei Mäusen rückgängig, die bereits erkrankt waren.“
Die Progeria-Mäuse entwickeln eine Herzkrankheit, die der von Kindern mit Progeria ähnelt. Die Autoren fanden heraus, dass der FTI sowohl die Entwicklung der Herzkrankheit bis zu einem gewissen Grad verhindern konnte, wenn die Mäuse ab dem Zeitpunkt ihrer Entwöhnung behandelt wurden, als auch die bereits bestehende Krankheit teilweise rückgängig machen konnte, wenn die Mäuse ab einem Alter von 9 Monaten behandelt wurden. „Aus meiner Sicht war eines der bemerkenswertesten Dinge die Fähigkeit, die Krankheit rückgängig zu machen“, sagte Collins, was entscheidend ist, da Progeria im Allgemeinen nicht bei der Geburt diagnostiziert wird, sondern erst, wenn Kinder Symptome zeigen und ein Teil des Schadens bereits angerichtet ist.
„Wenn sich herausstellt, dass diese Medikamente bei Kindern ähnliche Wirkungen haben, könnte dies einen großen Durchbruch bei der Behandlung dieser verheerenden Krankheit bedeuten“, sagte Dr. Nabel vom NHLBI, einer der Mitautoren der Studie. „Darüber hinaus werfen diese Erkenntnisse Licht auf die potenzielle Rolle von FTI-Medikamenten bei der Behandlung anderer Formen der koronaren Herzkrankheit.“
Den Artikel finden Sie in Wissenschaftlicher Amerikaner, „Neue Hoffnung für Progerie: Medikament für seltene Alterskrankheit“, bei https://www.sciam.com/article.cfm?id=new-hope-for-progeria-drug-for-rare-aging-disease und die Pressemitteilung des NIH unter https://www.nih.gov/news/health/oct2008/nhgri-06.htm
* Capell et al., „Ein Farnesyltransferase-Hemmer verhindert sowohl den Beginn als auch das späte Fortschreiten einer Herz-Kreislauf-Erkrankung in einem Progeria-Mausmodell.“ Verfahren der Nationalen Akademie der Wissenschaften, Band 105, Nr. 41, 15902-15907 (14. Oktober 2008)
In einer zweiten Studie, die online im Journal of Medical Genetics** veröffentlicht wurde, schuf das Forschungsteam von Dr. Maria Eriksson am Karolinska Institutet in Schweden ein weiteres Mausmodell für Progerie mit Haut- und Zahnveränderungen. Die Mäuse sind genetisch so verändert, dass die Progerie-Mutation jederzeit abgeschaltet werden kann. Sobald die Krankheit sichtbar wurde, wurde das Gen für Progerie abgeschaltet. Nach 13 Wochen war die Haut von normaler Haut kaum noch zu unterscheiden. Diese Studie zeigt, dass die Expression der Progerie-Mutation in diesen Geweben keine irreversiblen Schäden verursacht und dass eine Umkehrung der Krankheit möglich ist, was vielversprechende Ansätze für eine Behandlung von Progerie bietet.
**Eriksson et al., „Reversibler Phänotyp in einem Mausmodell des Hutchinson-Gilford-Progerie-Syndroms.“ J. Med. Genet. online veröffentlicht am 15. August 2008; doi:10.1136/jmg.2008.060772
Um diesen Artikel zu kaufen, gehen Sie zu: https://jmg.bmj.com/cgi/rapidpdf/jmg.2008.060772v1
Weitere Belege für den Zusammenhang zwischen Progerie, normalem Alterungsprozess und Herzerkrankungen
Diese spannenden Studien von Capell und Eriksson zeigen, dass diese Ergebnisse über Progerie hinaus das Potenzial haben, allen Patienten mit Herz-Kreislauf-Erkrankungen zu helfen. Forscher haben entdeckt, dass das für Progerie verantwortliche toxische Protein tatsächlich in geringen Mengen bei allen Menschen produziert wird und sich möglicherweise mit zunehmendem Alter ansammelt. Durch die Untersuchung dieser seltenen Kinder können wir also unser Verständnis eines wichtigen Mechanismus des menschlichen Alterns erweitern – und vielleicht neue Wege finden, den Prozess zu verlangsamen.
Frühjahr 2007: Höhepunkte des wissenschaftlichen Workshops der Progeria Research Foundation 2007: Fortschritte in der translationalen Wissenschaft
2006: Progerie 101/FAQs
2004: Genmutation verursacht fortschreitende Veränderungen der Zellstruktur bei Kindern mit Progerie Genmutation verursacht fortschreitende Veränderungen der Zellstruktur bei Kindern mit Progerie
2003: Identifizierung eines Gens gibt Kindern mit Progerie Hoffnung
Melden Sie sich an
für unsere
Neuigkeiten!
Gemeinsam
WILLE
Finde die Heilung!
