À propos de la progéria
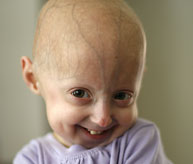 Le syndrome de Hutchinson-Gilford-Progeria (« progéria » ou « HGPS ») est une maladie génétique rare et mortelle caractérisée par un vieillissement accéléré chez les enfants. Son nom est dérivé du grec et signifie « prématurément vieux ». Bien qu’il existe différentes formes de progéria*, le type classique est le syndrome de Hutchinson-Gilford-Progeria, qui doit son nom aux médecins qui l’ont décrit pour la première fois en Angleterre, en 1886 par le Dr Jonathan Hutchinson et en 1897 par le Dr Hastings Gilford.
Le syndrome de Hutchinson-Gilford-Progeria (« progéria » ou « HGPS ») est une maladie génétique rare et mortelle caractérisée par un vieillissement accéléré chez les enfants. Son nom est dérivé du grec et signifie « prématurément vieux ». Bien qu’il existe différentes formes de progéria*, le type classique est le syndrome de Hutchinson-Gilford-Progeria, qui doit son nom aux médecins qui l’ont décrit pour la première fois en Angleterre, en 1886 par le Dr Jonathan Hutchinson et en 1897 par le Dr Hastings Gilford.
Le syndrome de Heidegger est causé par une mutation du gène LMNA (prononcé lamin-a). Le gène LMNA produit la protéine Lamin A, qui est l’échafaudage structurel qui maintient le noyau d’une cellule. Les chercheurs pensent désormais que la protéine Lamin A défectueuse rend le noyau instable. Cette instabilité cellulaire semble conduire au processus de vieillissement prématuré de la progéria.
Bien qu'ils naissent en bonne santé, les enfants atteints de progéria commencent à présenter de nombreuses caractéristiques de vieillissement accéléré au cours des deux premières années de vie. Les signes de la progéria comprennent un retard de croissance, une perte de graisse corporelle et de cheveux, une peau vieillissante, une raideur des articulations, une luxation de la hanche, une athérosclérose généralisée, une maladie cardiovasculaire (cardiaque) et un accident vasculaire cérébral. Les enfants ont une apparence remarquablement similaire, malgré des origines ethniques différentes. Sans traitement, les enfants atteints de progéria meurent d'athérosclérose (maladie cardiaque) à un âge moyen de 14,5 ans.
* D'autres syndromes progéroïdes incluent le syndrome de Werner, également connu sous le nom de « progéria de l'adulte », qui n'apparaît qu'à la fin de l'adolescence et dure jusqu'à 40 et 50 ans.
S'inscrire
pour notre
Mises à jour !
Ensemble, nous
VOLONTÉ
trouve le remède !
